
![]()
CRISPR GENE EDITING
Editas Medicine is a leading gene editing company dedicated to developing a robust pipeline of medicines to treat people living with serious diseases around the world.
We have built a platform that utilizes CRISPR gene editing, a revolutionary approach to developing medicines. Advances in this technology have made it possible to modify almost any gene in human cells—meaning we may soon be able to treat a wider range of diseases.
Gene editing is a complicated topic. To understand how gene editing can fit into the larger picture of making medicines, we believe it helps to start with some basics of genetics. If you are already familiar and just want to know how Editas Medicine is specifically using gene editing, jump to that section. Otherwise, let’s start with the basics.
Genetics
Genes, Proteins, and DNA
The human body is made up of trillions of cells.
Each cell has a copy of your genome—the total collection of all your genes and DNA.
Genes are instructions that every cell in your body uses to make the proteins they need to function.
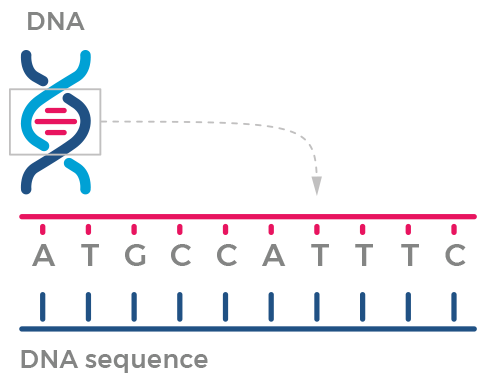
Genes can be found in long chains of molecules called deoxyribonucleic acids (DNA) that are twisted together into the shape of a double helix.
There are 4 DNA molecules that are identified by the letters A, T, C, and G. Combinations of these letters make up the genetic instructions that our cells use to make proteins.
DNA Mutations and Disease
Our genes can also be a source of disease.
Small breaks in our DNA are incredibly common and are normally uneventful. DNA breaks can happen from sunlight, for example, or during cell divisions that happen as we grow. Our cells have built-in DNA repair processes that constantly fix these breaks as they occur.
However, breaks are sometimes repaired incorrectly, creating what is known as a mutation. Mutations can occur spontaneously or be passed down from our parents. Mutations can also change how our cells function, and may lead to serious diseases such as sickle cell disease (SCD), Leber congenital amaurosis 10 (LCA10), cancer, and many others.
Genomic Medicine: Revolutionizing the Development of Medicines
Genomic medicine is a developing field of medical research that harnesses recent advances in genetics to develop new medicines for diseases. In fact, we are at an inflection point where we are seeing the first real genomic medicines emerge to help patients.
Advances in the scientific fields of cell therapy and gene therapy have paved the way for the success of gene editing, which in turn is now expanding and accelerating the development of a new class of genomic medicines.

This field genetically addresses diseases by using gene editing and gene therapy technologies, which are different in a few key ways.
In gene editing, a mutated gene is revised, removed, or replaced at the DNA level. In gene therapy, the effect of a mutation is offset by inserting a “healthy” version of the gene, and the disease-related genes remain in the genome. Both approaches may provide a durable benefit to patients, and both gene therapy and gene editing, alone or in combination, may lend themselves to the development of transformative genomic medicines.
Editas Medicine is working to develop genomic medicines for a range of diseases, many of which do not currently have medicines available. We are a gene editing company, and use the terms “gene” and “genome” editing interchangeably to refer to our CRISPR gene editing technology.

CRISPR Gene Editing
CRISPR (pronounced “crisper”) is an acronym for “Clustered, Regularly Interspaced, Short Palindromic Repeats,” and refers to a recently developed gene editing technology that can revise, remove, and replace DNA in a highly targeted manner. CRISPR is a dynamic, versatile tool that allows us to get to and edit nearly any location in the genome, and has the potential to help us develop medicines for people with a wide variety of diseases. We view CRISPR as a “platform” technology because of its ability to target DNA in any cell or tissue.
CRISPR uses a combination of 2 types of molecules to edit disease-related genes or to modify cells: a nuclease (the gene editor) and guide RNA (which helps the nuclease find the right place to edit).
CRISPR’s ability to only edit intended DNA targets and avoid off-target editing is known as its specificity. Achieving high levels of specificity requires the right combination of nuclease and guide RNA.
Nuclease
Protein that edits DNA
Editas Medicine works with CRISPR nucleases, Cas9 (both aureus Cas9 and pyogenes Cas9) and Cas12a, including enhanced forms of each.
Guide RNA
Molecule that shows the nuclease where to edit
Editas Medicine creates and tests thousands of guide RNAs to identify guides with the highest levels of specificity.
About the Cas9 Nuclease
Cas9 is able to locate, bind to, and edit targeted genes. It is an essential tool for the research and development of genomic medicines and has the potential to facilitate solutions for many diseases.
About the Cas12a (Cpf1) Nuclease
Cas12a—also known as Cpf1—greatly expands the range of ways that CRISPR gene editing can be used to produce genomic medicines. It provides researchers with more options for where and how to edit DNA and makes use of a shorter guide RNA than Cas9. Our team is continuing to evaluate the best uses for each nuclease.
SLEEK Gene Editing
SLEEK gene editing is an optimized approach to developing the next generation of cell therapy medicines for cancer and other serious diseases. Utilizing our proprietary engineered AsCas12a nuclease, SLEEK enables high efficiency, multi-transgene knock-in of induced pluripotent stem cells (iPSCs), T cells, and natural killer (NK) cells while ensuring robust, transgene expression.
![]()
UDiTaS™
UDiTaS™ is a methodology developed by Editas Medicine for simultaneous measurement of on-target editing, off-target editing, large insertions, deletions, translocations, and other structural changes. The method improves on previous uni-directional amplification techniques (eg: AMP-Seq and HTGTS), implementing a robust and rapid tagmentation step with a custom designed Tn5 transposase which includes unique molecular identifiers (UMIs) for better quantitation of these events.
We’re Developing Many Gene Editing Medicines
Using CRISPR gene editing technology, we are pioneering the possible and developing transformative genomic medicines for people living with serious diseases.
We are working on making medicines where editing occurs inside the body (in vivo gene editing medicines).

Editas Medicine’s In Vivo Gene Editing Medicines Advances
We’re using in vivo technology to develop a wide range of gene editing medicines, focusing on hematopoietic stem cells and other tissue types.
Currently developing next generation in vivo medicines, including in vivo editing of hematopoietic stem cells (HSCs), liver, and other tissues.
These programs are currently in the preclinical phase.
In January 2025, Editas announced new in vivo preclinical proof of concept data of hematopoietic stem and progenitor cell (HSPC) editing and fetal hemoglobin (HbF) induction in non-human primates.
In this initial non-human primate (NHP) study, we achieved effective delivery to 75% of hematopoietic stem cells (HSCs) and meaningful levels of HSC editing of 17% with Editas’ proprietary targeted lipid nanoparticles (tLNPs) after a single dose of tLNP. We will continue to evaluate and further optimize LNP formulations to achieve therapeutic editing levels.
In addition, we achieved NHP proof of concept validating high efficiency gene editing in the liver with the first use of AsCas12a delivery by LNP and demonstrated proof of upregulation strategy in mice by increasing the clinically relevant target protein resulting in a significant disease biomarker reduction for an undisclosed liver target.
In October 2024, Editas announced in vivo preclinical proof of concept of HSPC editing and HbF induction in humanized mice engrafted with human HSCs and lacking their own hematopoietic cells.
We observed high levels of editing of the HBG1/2 promoter, leveraging our clinically validated upregulation strategy, utilizing a novel and Editas-proprietary tLNP formulation for non-liver or extrahepatic tissue delivery.
We have also achieved an editing level of 29% in HSPCs after a single dose by utilizing an HSC targeting strategy and tLNP formulation optimization.
The preclinical data further demonstrated that editing with the Company’s proprietary tLNP formulation resulted in the functional outcome of HbF induction, indicated by the presence of HbF expressing human red blood cells (on average 20%) that populate in the host by one month.
Editas Medicine is developing CRISPR medicines for 2 major blood diseases: SCD and beta-thalassemia.
Sickle Cell Disease
- A disease that affects the shape and function of the body’s red blood cells, the cells that carry oxygen throughout the body
- In SCD, the red blood cells are misshapen, in a sickle shape instead of the disc shape. The abnormal shape causes the cells to block blood flow causing pain and early death
- There are an estimated 100,000 people in the United States currently living with SCD
- Usually begins in early childhood. Early symptoms include:
- Low red blood cell counts
- Infections
- Pain
- Fatigue
- Delayed growth and development
- The current standard of care is to manage symptoms and complications; there are currently no approved treatments for SCD
Beta thalassemia
- A blood disorder that reduces the body’s production of hemoglobin, a protein that carries oxygen to all of the body’s cells
- Classified into 2 types based on severity of the disorder:
- Thalassemia major (more severe; also known as Cooley’s anemia)
- Thalassemia intermedia (less severe)
- Thousands of babies around the world are born with this disorder each year
- The symptoms and age where they start are different for each type of beta-thalassemia. General symptoms include:
- Low red blood cell counts and anemia
- Growth problems
- Bone abnormalities
- In more severe cases: yellowing of the skin and eyes and delayed puberty
Editas Medicine’s In Vivo Gene Editing Medicines Advances
We are partnering with others to use gene editing to develop medicines.
Advancing cellular therapy assets through partnerships, including continued development of alpha-beta T-cells medicines with Bristol Myers Squibb and gamma-delta T-cell medicines with Immatics.